Overview
Accurate and comprehensive annotation of transcript sequences is essential for transcript quantification as well as differential gene, and transcript expression analysis. The magnitude and dynamics of transcription and post-transcriptional reprogramming of the transcriptome provide insights into the cellular complexity of external and internal cue responses. Traditional short-read RNA-seq has been widely used to characterize transcript and gene expression changes. While this approach is highly effective in quantifying transcript abundance, short-read segments (typically 100 to 250 base pairs) rarely span full-length transcripts, which can often be several thousand bases long, making it difficult to directly infer full-length transcript structure. These limitations are particularly evident in the complex human transcriptome. The landscape of transcriptomic analysis has undergone significant advancements with the introduction of non-comparison quantification tools, namely Kallisto and Salmon. These platforms have markedly transformed the quantification paradigm of individual transcript expression levels, especially when utilizing Illumina short-read RNA-seq data. A critical consideration, however, when using Kallisto and Salmon, is the absolute requirement of a reference transcriptome. The fidelity and accuracy of transcript quantification hinge substantially on the quality, granularity, and all-encompassing nature of the selected reference transcriptome. In essence, a sub-optimal or incomplete reference can adversely influence the outcome of the analysis.
Transitioning into a new epoch of RNA-seq, the embrace of long-read PacBio Single Molecule, Real-Time (SMRT) sequencing technology has provided unprecedented depth and clarity. This method, colloquially termed the Iso-Seq approach, has enabled the capture of extensive sequencing reads, with documented lengths reaching up to an impressive 60 kilobases. Furthermore, this technique brings forth enhanced structural coherence of the transcribed sequences. What sets the Iso-Seq method apart is its unrivaled ability to conduct full-length isoform RNA sequencing, offering researchers the latitude to delve into comprehensive transcriptomic landscapes or conduct an in-depth examination of specific gene entities in a more targeted fashion.
The inherent strengths of SMRT sequencing lie in its superior detection capabilities. It has an unparalleled proficiency in pinpointing transcription start and end points, categorized as TSS (Transcription Start Sites) and TES (Transcription End Sites) respectively. Moreover, it provides an acute understanding of alternative splicing (AS) dynamics, sheds light on alternative polyadenylation (APA) events, and most importantly, ensures an accurate alignment of varied combinations of TSS, TES, and splice junctions (SJs). This intricate ability to discern these unique transcriptomic events ensures that the analyses conducted using SMRT sequencing are of the highest resolution and precision.
CD Genomics offers specialized full-length transcript sequencing (Iso-Seq) services. With long, accurate HiFi reads, you can characterize the complete diversity of the transcriptome - up to tens of bases. However, long-read sequencing has its own limitations, such as the inability to accurately quantify gene expression because the throughput of long-read platforms is relatively low compared to short-read methods.
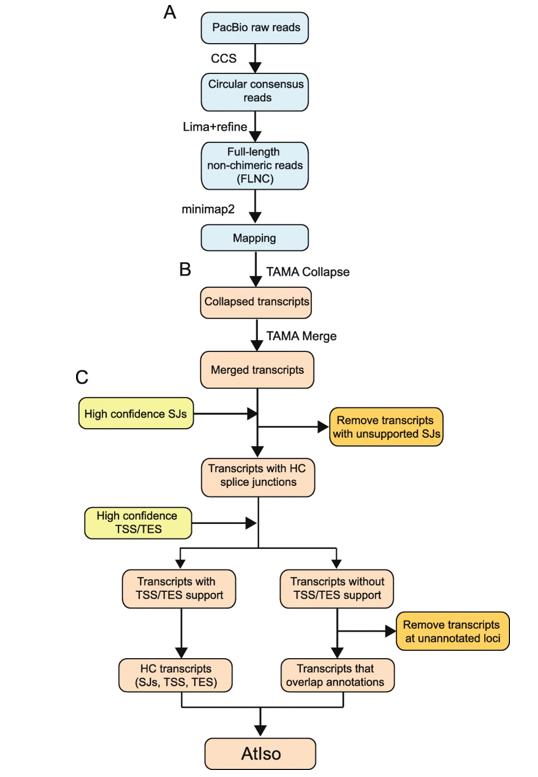 Workflow of analysis of PacBio Iso-sequencing. (Zhang et al., 2022)
Workflow of analysis of PacBio Iso-sequencing. (Zhang et al., 2022)
What is Iso-Seq?
The Isoform Sequencing (Iso-Seq) method is an intricate protocol designed to leverage the capabilities of the Pacific Biosciences SMRT sequencing technology for the purpose of sequencing full-length complementary DNA (cDNA). This strategy offers a considerable advantage over other techniques primarily because it mitigates the need for fragment assembly, which can introduce errors and complexities in downstream analyses.
Advantages of Iso-Seq
- Full-length transcripts: One of the most significant advantages of the Iso-Seq method is its ability to generate full-length reads. This eliminates the need to piece together short reads, simplifies the process of isoform determination, and reduces the possibility of assembly errors.
- Insight into selective splicing: In eukaryotes, genes are often selectively spliced, resulting in multiple transcript variants of a single gene. Iso-Seq provides a window into this complex world, providing a clearer understanding of splicing events and their functional implications.
- Improved genome annotation: By generating long, continuous reads, Iso-Seq facilitates accurate annotation of genomes. Researchers use it to discover new genes and correct previous annotations, even in widely studied organisms.
- Versatility: The Iso-Seq method is not limited to eukaryotes, but is also applicable to prokaryotic systems, leading to fine annotation and new discoveries.
- For species without a reference genome: The Iso-Seq bioinformatics analysis workflow does not require a reference genome, although if a reference genome is available, it can be used to map the full-length transcripts back to the genome.
Library Preparation and Extraction of Read-Of-Insert From Pacbio Iso-Seq
Depending on the end goal, these sequencing libraries are constructed using a variety of kits such as the Clontech SMARTer PCR kit. The length of the resulting sequencing reads is influenced by the quality of the RNA and the successful generation of full-length cDNA.
To enhance the expression of full-length cDNAs, cap-dependent junctions can be used or Poly(A)+ RNA selection can be combined with 5' capped mRNA capture. These full-length mRNAs serve as templates for cDNA synthesis followed by size selection.
A major innovation of PacBio's new Sequel System is the ability to sequence cDNAs without prior size selection. This process results in an SMRTbellTM library that can be sequenced on either the RSII or Sequel platform. This approach ensures full-length cDNA sequencing with minimal loss of sequence ends.
At the heart of the PacBio sequencing strategy is the utilization of Zero Mode Waveguide (ZMW) technology, which consists of nanopores that hold sequencing templates. When fluorescently labeled DNA bases are incorporated, they emit a signal that is captured in real-time. Hairpin junctions added to the DNA during library preparation create circular DNA templates. This circularity allows the polymerase to pass through the template multiple times, thus improving sequencing accuracy.
After sequencing, the bioinformatics workflow involves converting the raw data into actionable insights. Tools and pipelines such as SMRT Link extract valid sub-reads and then extract ROIs for each ZMW. The ToFu PacBio pipeline plays a critical role in extracting ROIs and full-length non-chimeric (FLNC) reads. These reads were further refined using iterative clustering to ensure high consensus accuracy.
Applications of Iso-Seq
Crop Improvement and Agriculture
One of the most important applications of Iso-Seq methods is in agriculture. Given the growing global demand for food, improving crop yields and resistance has become critical. Iso-Seq plays a key role in exploring the transcriptomes of a wide range of crops, including maize, wheat, rice, and grapes. By providing insights into diverse and complex transcriptomes, PacBio is paving the way for a better understanding of the factors that influence yield, disease resistance, and environmental stress response.
Oncology and Fusion Gene Detection
The Iso-Seq method significantly enhances cancer research. Its ability to detect fusion genes has proven invaluable, especially when these genes play a key role in tumorigenesis. For example, the detection of the IGH-DUX4 fusion in B-cell acute lymphoblastic leukemia demonstrates the clinical relevance and potential therapeutic implications of this technology.
The cellular heterogeneity present in tissues, especially in complex organs, is often lost when bulk sequencing is performed. PacBio's Iso-Seq method is customized for single-cell studies and can uncover cell type-specific subtypes. This has led to groundbreaking discoveries, particularly in the field of neurobiology, where unique isoforms have been found in postnatal mouse brains and Down syndrome aging brains.
Differential Expression and Subtype Analysis
One area where Iso-Seq methods really come into play is differential expression analysis. It uniquely identifies differential isoform usage (DIU) while aligning with the gene level expression of short-read data. This nuanced understanding is not possible with traditional methods and provides a clearer picture of gene regulation and expression patterns.
Predicting Full-length Open Reading Frames
A comprehensive understanding of open reading frames (ORFs) is essential for functional genomics. The Iso-Seq method enables the sequencing of full-length cDNAs, providing a clear picture of the ORF. This facilitates accurate protein prediction, contributing to proteomics studies and ensuring that our annotations reflect the true coding potential of the genome.
Alternative Start and End Site Detection
One of the fundamental applications of the Iso-Seq method is its ability to accurately detect alternative transcription start and end sites. Conventional short-read-long sequencing methods often struggle to capture the full diversity of transcriptional isoforms, especially those with different ends. With Iso-Seq, researchers can obtain full-length reads covering the entire transcript, revealing alternative start and stop sites with unrivaled precision.
Integrated Splicing Event Characterization
Selective splicing is a major source of protein diversity in eukaryotes. By harnessing the power of long read lengths, Iso-Seq can capture and characterize complex splicing patterns in the transcriptome. This provides insights into gene regulatory mechanisms and can reveal key variants associated with disease or developmental stages.
Mining Non-coding RNA
Non-coding RNAs play key roles in various cellular processes. However, their full identification and characterization remains challenging. With PacBio's Iso-Seq technology, researchers can now discover and annotate non-coding RNAs, enhancing our understanding of their functional significance in health and disease.
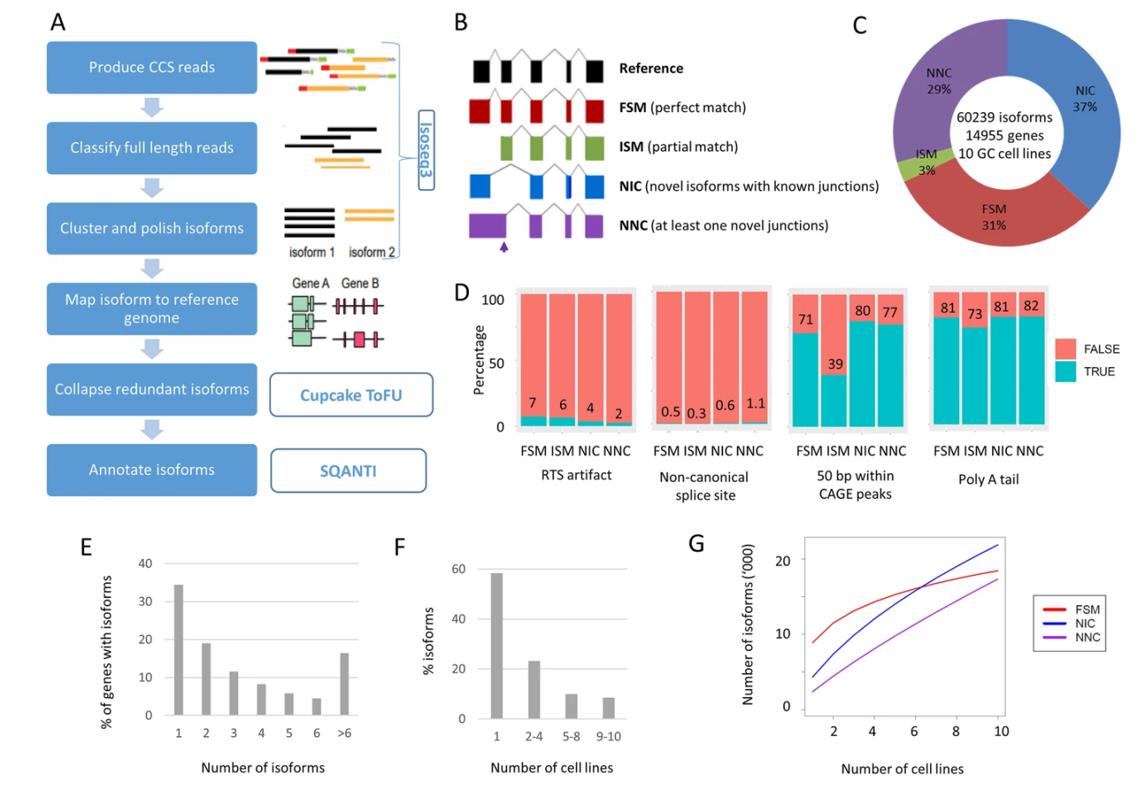 Landscape of long-read transcriptome in gastric cancer cell lines. (Huang et al., 2021)
Landscape of long-read transcriptome in gastric cancer cell lines. (Huang et al., 2021)
References
- Zhang, Runxuan, et al. "A high-resolution single-molecule sequencing-based Arabidopsis transcriptome using novel methods of Iso-seq analysis." Genome biology. 23.1 (2022): 149.
- Huang, Kie Kyon, et al. "Long-read transcriptome sequencing reveals abundant promoter diversity in distinct molecular subtypes of gastric cancer." Genome biology. 22 (2021): 1-24.